今日热闻!标记基因是什么意思?标记基因筛选原理是什么?
标记基因(标记基因筛选原理)
Knight & Yang-Yu Liu. (2021). Challenges in benchmarking metagenomic profilers. Nature Methods, doi: https://doi.org/10.1038/s41592-021-01141-3
 (资料图片仅供参考)
(资料图片仅供参考)
随着越来越多的研究揭示出微生物组与人体健康的密切关系,宏基因组测序尤其是全宏基因组鸟枪法测序(whole metagenome sequencing,WMS)作为微生物组学最重要的研究手段之一被学术界、工业界广泛使用。为了解读高通量WMS数据,许多用于物种分类的生物信息学工具被开发出来,而这其中能够避免拼接等繁重计算任务的MetaPhlAn、Kraken、PathSeq等在大量宏基因组研究种被应用。但是目前在正确评价和使用这些生信工具以及解读相应的输出结果方面并没有引起足够的重视。比如,不同工具的输出结果之间具有很大的差异,研究人员往往将其归因于不同工具所用数据库的差别。但是我们发现,不同生信工具输出的“丰度类型”存在根本性的差别,是生信工具之间分析结果差异产生的本质原因之一。忽视和混淆这一丰度类型的差别,将改变生信工具性能评价的结果,并深刻影响对宏基因组测序数据的解读。另外,该问题也会严重阻碍荟萃研究,影响跨研究之间结果的可比性,并导致微生物组研究在临床医学转化上的困难。
2021年5月13日,哈佛大学医学院刘洋彧团队与加州大学圣地亚哥分校Rob Knight团队在Nature Methods上发表了题为Challenges in Benchmarking Metagenomic Profilers的论文。该研究通过数据模拟,对宏基因组物种分类工具的输出结果进行了深度解读,创造性的提出了基于不同丰度类型(基于序列或基于物种分类相对丰度)的双重评价标准,为解决微生物组研究中如何选择宏基因组学物种分类工具的问题提供了重要依据,也对微生物组标准化研究提出了一系列建设性的意见。
模式图:基于物种分类(标记基因,如MetaPhlAn2)和基于序列方法(如Kraken2)对物种分类定量产生巨大差异,主要受微生物基因组大小影响。
在宏基因组测序分析中,序列(sequence)丰度和物种(taxonomic)丰度是两种截然不同的相对丰度类型。前者序列(sequence)丰度是计算属于某一物种经过测序后的DNA在整个菌群DNA中的百分比,而后者物种(taxonomic)丰度则代表某一物种的个体数量在菌群总个体数中的百分比。宏基因组学物种分类工具可根据其使用数据库的类型而分为三类:DNA-to-DNA,DNA-to-Protein,DNA-to-Marker。通过设计一个简单的模拟菌群,我们发现不同类型工具输出的相对丰度类型并不统一,比如DNA-to-DNA方法的(代表软件Kraken和Bracken)输出丰度类型为序列丰度,而DNA-to-Marker方法的(代表软件MetaPhlAn和mOTUs)输出的丰度类型为物种丰度(如下图1所示)。
图1. 三种物种定量方法的比较。a. 模式图;b. 两种基因组的模拟群落;c. 不同软件定量的结果。
通过模拟数据,研究人员将序列丰度和物种丰度分别作为金标准,对不同的宏基因组学物种分类工具进行评价,结果发现,在以序列丰度为金标准时,DNA-to-DNA方法的表现优于DNA-to-Marker方法,而在以物种丰度为金标准时,结果则相反。因此,物种分类软件的表现与测评时作为金标准的相对丰度类型有很大关系。
混淆序列丰度与物种丰度会对宏基因组数据的解读产生四个方面的重要影响:
1. 在解析物种构成方面:如果使用序列丰度作为解读标准,将高估大基因组物种并且低估小基因组物种在菌群中的真实数量。在复杂的菌群中,微生物基因组的大小存在很大的差别,只在细菌内部,理论上基因组的差别就可以达到100倍,而跨物种(如病毒和真菌)微生物基因组的差别更无法估量。理解序列丰度和物种丰度,对临床诊断病原菌过程中如何设置阈值十分关键。
2. 在alpha多样性方面:与使用物种丰度相比较,如果使用序列丰度作为解读标准,将会整体上降低样本的alpha多样性(Shannon, Simpson and Pielou’s evenness index),但这一改变并不是严格一致的,部分样本的alpha多样性反而会升高。在当前宏基因组研究受样本量局限的情况下,这将会导致微生物样本alpha多样性的排序混乱,进而影响到alpha多样性在个体和组间比较的一致性和可重复性。
3. 在beta多样性方面:通过设计模拟菌群,我们基于不同beta多样性分析方法(BC,rJSD,L1,L2,rAD)比较了以两种不同相对丰度为基础的样本间关系,通过检验我们发现序列丰度所描述的样本间关系与物种丰度所描述的样本间关系存在差别,相关性为0.51-0.94。因此,以不同生信工具输出结果为下游分析起点,可能得到不同的样本间或组间关系。
4. 在排列分析(ordination analysis)方面:排列分析是宏基因组常用的分析手段,通过将N维的物种构成数据降低到两维或者三维来比较和展示个体或组间的差异。对于同一批样本,基于序列丰度和基于物种丰度的排列分析所产生的结果相差很大,无论是NDMS, PCoA, t-SNE或UMAP方法所产生的二维散点图,其经过一致性分析后,都表现出很大的差异性。也就是说,在基于不同生信工具所产生的下游分析中,有可能发生组间差异无法重复的情况。
本文通过严谨的论证分析,量化了宏基因组学物种分类工具所产生的两种相对丰度类型的差别,对于混淆两种丰度所产生的影响进行了全面系统地研究。由于存在大量未知微生物基因组和多倍体信息缺失等原因,将物种丰度与序列丰度之间进行转换存在现实难度,往往无法达到预期目标,因此选择合适的宏基因组学物种分类工具十分关键。目前无论是DNA-to-DNA方法(以Kraken为代表,产生序列丰度)还是DNA-to-Marker方法(以MetaPhlAn为代表,产生物种丰度),都是宏基因组研究中的重要工具,并且已经被应用于大量研究中。虽然在方法一致的前提下,丰度的差别不会影响到同一个实验中组间的比较,但这不可避免地影响了诸多已发表的微生物组相关研究结论的可解读性,也将为回顾性的荟萃分析带来极大的挑战。因此我们呼吁微生物领域研究人员审慎解读宏基因组测序结果,严格区分相对丰度类型,重新审视过往基于序列丰度的研究结论。鉴于物种丰度更具生物学和生态学意义,我们也建议大家开发更多基于DNA-to-Marker方法的宏基因组学物种分类工具。
本文第一作者是哈佛大学医学院的孙政博士和加州大学圣地亚哥分校的黄适博士。Rob Knight教授和刘洋彧教授为本文的通讯作者。
图2. 对不同界的物种对序列和分类两种定量结果的相关分析
图3. 使用Bracken、Kraken2、mOTUs2和MetaPhlAn2共4种软件对模拟群落不同估计方法定量结果的评测。
图4. 基于序列和物种丰度计算Alpha多样性
图5. 对两种定量方法结果在不同样本类型上的排序分析。
- 今日热闻!标记基因是什么意思?标记基因筛选原理是什么? 标记基因(标记基因筛选原理)Knight&Yang-YuLiu (2021) Challengesinbenchmarkingmetagenomicprofilers NatureMethods,do
-
 当前热点-萨德是什么?为什么萨德事件平息了? 萨德是什么(为什么萨德事件平息了)美国遏制中国并不仅仅是从贸易战开始,并不仅仅是从科技战开始,也不仅仅是从特朗普开始,它实际上早...
当前热点-萨德是什么?为什么萨德事件平息了? 萨德是什么(为什么萨德事件平息了)美国遏制中国并不仅仅是从贸易战开始,并不仅仅是从科技战开始,也不仅仅是从特朗普开始,它实际上早... -
 要闻:有什么小说比较好看?十本最好看的小说推荐 有什么小说好看(十本最好看的小说推荐)小s,每天给你带来精品小说推荐!今天,给大家推荐十本不小白不烂尾的完本小说推荐,爽点满满,值...
要闻:有什么小说比较好看?十本最好看的小说推荐 有什么小说好看(十本最好看的小说推荐)小s,每天给你带来精品小说推荐!今天,给大家推荐十本不小白不烂尾的完本小说推荐,爽点满满,值... -
 【热闻】今日南阳油田价格是多少?南阳油田居民网
【热闻】今日南阳油田价格是多少?南阳油田居民网 -
 每日速递:七十二地煞术是什么?地煞七十二术全解 七十二地煞术(地煞七十二术全解)少年时读西游记,听到孙悟空会地煞七十二变一直以为是可以变化成72种东西,长大后才了解到七十二变可...
每日速递:七十二地煞术是什么?地煞七十二术全解 七十二地煞术(地煞七十二术全解)少年时读西游记,听到孙悟空会地煞七十二变一直以为是可以变化成72种东西,长大后才了解到七十二变可... -
 每日关注!雪基龙牛仔裤好看吗?直筒牛仔裤推荐 雪基龙牛仔裤(直筒牛仔裤)范丞丞《时尚COSMO》七月刊单人图集,“选择练习选择勇敢的自己” 摄影:@于聪YuCong昨日,@宋威龙99现身...
每日关注!雪基龙牛仔裤好看吗?直筒牛仔裤推荐 雪基龙牛仔裤(直筒牛仔裤)范丞丞《时尚COSMO》七月刊单人图集,“选择练习选择勇敢的自己” 摄影:@于聪YuCong昨日,@宋威龙99现身...
-
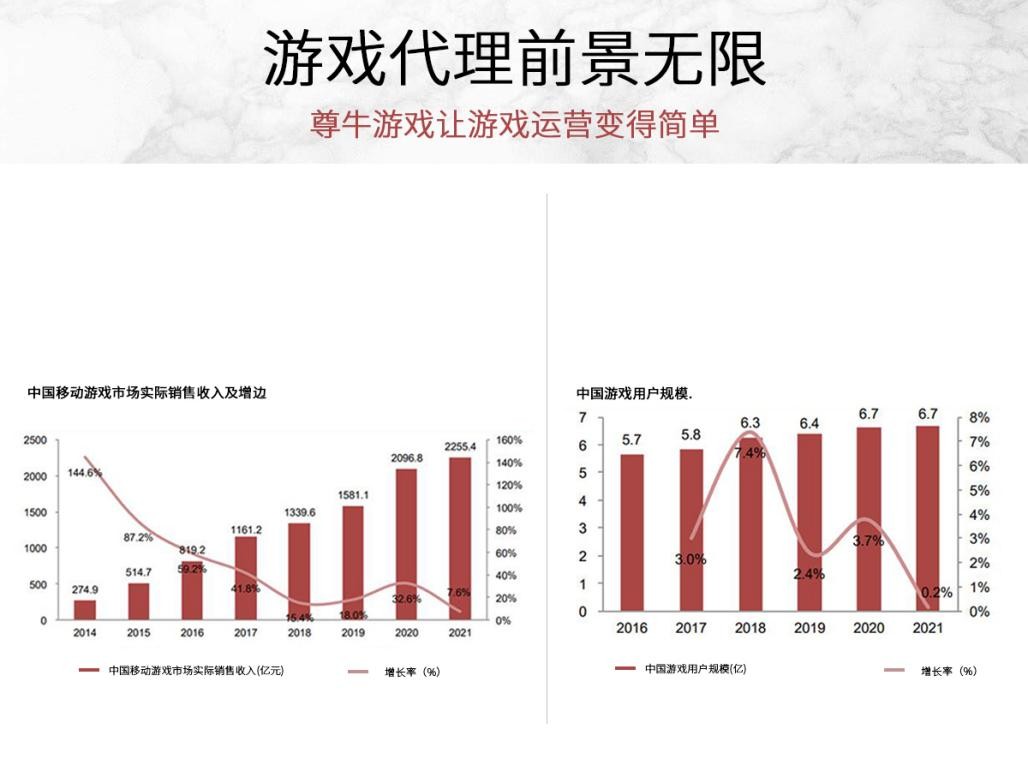 游戏也能赚钱 手游代理从尊牛游戏起航 游戏在我们生活中占据越来越重要的地位,据统计,2021年我国游戏用户达到6 7亿人,占总人口的一半数量以上,与此同时游戏产业销售额为2965
游戏也能赚钱 手游代理从尊牛游戏起航 游戏在我们生活中占据越来越重要的地位,据统计,2021年我国游戏用户达到6 7亿人,占总人口的一半数量以上,与此同时游戏产业销售额为2965 - 【热闻】今日南阳油田价格是多少?南阳油田居民网
-
 【独家】屈原的故事是什么?端午节真正的的由来和习俗是什么? 屈原的故事端午节来历(端午节真正的的由来和习俗)端午节是我国古老的传统节日,始于春秋战国时期,已有着两千多年的历史。每逢这个节...
【独家】屈原的故事是什么?端午节真正的的由来和习俗是什么? 屈原的故事端午节来历(端午节真正的的由来和习俗)端午节是我国古老的传统节日,始于春秋战国时期,已有着两千多年的历史。每逢这个节... - 要闻:有什么小说比较好看?十本最好看的小说推荐 有什么小说好看(十本最好看的小说推荐)小s,每天给你带来精品小说推荐!今天,给大家推荐十本不小白不烂尾的完本小说推荐,爽点满满,值...
-
 当前热点-st博元股吧:600656重组上市消息 st博元股吧(600656重组上市消息)===本文导读===*ST博元涨停“告别”散户赌“老八股”不死?*ST博元今天上演“告别秀”投资者表现淡定*...
当前热点-st博元股吧:600656重组上市消息 st博元股吧(600656重组上市消息)===本文导读===*ST博元涨停“告别”散户赌“老八股”不死?*ST博元今天上演“告别秀”投资者表现淡定*... - 当前热点-萨德是什么?为什么萨德事件平息了? 萨德是什么(为什么萨德事件平息了)美国遏制中国并不仅仅是从贸易战开始,并不仅仅是从科技战开始,也不仅仅是从特朗普开始,它实际上早...
- 每日速递:七十二地煞术是什么?地煞七十二术全解 七十二地煞术(地煞七十二术全解)少年时读西游记,听到孙悟空会地煞七十二变一直以为是可以变化成72种东西,长大后才了解到七十二变可...
- 每日关注!雪基龙牛仔裤好看吗?直筒牛仔裤推荐 雪基龙牛仔裤(直筒牛仔裤)范丞丞《时尚COSMO》七月刊单人图集,“选择练习选择勇敢的自己” 摄影:@于聪YuCong昨日,@宋威龙99现身...
-
 今日播报!金汉斯多少钱?石家庄金汉斯自助餐贵不贵? 金汉斯多少钱(石家庄金汉斯自助餐)据了解,有着8年历史的济南本土老牌自助餐饮连锁企业“金汉斯烤肉”近日推出了“一票制”供应惠民服务...
今日播报!金汉斯多少钱?石家庄金汉斯自助餐贵不贵? 金汉斯多少钱(石家庄金汉斯自助餐)据了解,有着8年历史的济南本土老牌自助餐饮连锁企业“金汉斯烤肉”近日推出了“一票制”供应惠民服务... - 今日热闻!标记基因是什么意思?标记基因筛选原理是什么? 标记基因(标记基因筛选原理)Knight&Yang-YuLiu (2021) Challengesinbenchmarkingmetagenomicprofilers NatureMethods,do
-
 每日速递:清华团队发现“神奇药水”,可将多能干细胞转变成全能干细胞
IT之家6月23日消息,据清华大学公众号消息,清华大学药学院丁胜团队以哺乳动物小鼠为主要研究对象,经过6年多攻关为再生领域“寻药”,...
每日速递:清华团队发现“神奇药水”,可将多能干细胞转变成全能干细胞
IT之家6月23日消息,据清华大学公众号消息,清华大学药学院丁胜团队以哺乳动物小鼠为主要研究对象,经过6年多攻关为再生领域“寻药”,... -
 快看:苹果确认iPad不再支持用作Home Hub
IT之家6月23日消息,苹果今天证实,在今年秋季推出iOS16、iPadOS16、macOSVentura和HomePod16正式版软件
快看:苹果确认iPad不再支持用作Home Hub
IT之家6月23日消息,苹果今天证实,在今年秋季推出iOS16、iPadOS16、macOSVentura和HomePod16正式版软件 -
 实时:6月22日广东新增本土无症状感染者3例 通报详情6月22日0-24时,广东全省新增本土无症状感染者3例,均为深圳报告。全省新增出院2例(均为境外输入),目前在院69例(境外输入68例);
实时:6月22日广东新增本土无症状感染者3例 通报详情6月22日0-24时,广东全省新增本土无症状感染者3例,均为深圳报告。全省新增出院2例(均为境外输入),目前在院69例(境外输入68例); -
 热推荐:俄罗斯用户已无法下载和升级 Win11 预览版更新
IT之家6月23日消息,本月,微软宣布大幅缩减在俄罗斯的业务,其发言人称,超过400名员工将被裁。此前央视新闻还报道称,微软已经停止了在俄罗
热推荐:俄罗斯用户已无法下载和升级 Win11 预览版更新
IT之家6月23日消息,本月,微软宣布大幅缩减在俄罗斯的业务,其发言人称,超过400名员工将被裁。此前央视新闻还报道称,微软已经停止了在俄罗 -
 今日快看!全新本田CR-V曝光:内饰彻彻底底变了 全新本田CR-V曝光:内饰彻彻底底变了
今日快看!全新本田CR-V曝光:内饰彻彻底底变了 全新本田CR-V曝光:内饰彻彻底底变了 -
 今日热讯:高通推出全新 AI 软件栈产品组合,让 AI 部署更轻松
IT之家6月22日消息,高通技术公司今日宣布推出高通AI软件栈产品组合,以提升高通在AI和智能网联边缘领域的领先优势。高通AI软件栈是面向O
今日热讯:高通推出全新 AI 软件栈产品组合,让 AI 部署更轻松
IT之家6月22日消息,高通技术公司今日宣布推出高通AI软件栈产品组合,以提升高通在AI和智能网联边缘领域的领先优势。高通AI软件栈是面向O -
 当前关注:肆意颁发“山寨证书” 这家被点名的“五假”公司仍在营业 近期,江苏英才职业技能鉴定有限责任公司(以下简称英才公司)以“JYPC全国职业资格考试认证中心”名义颁发各类“山寨证书”被人力资源...
当前关注:肆意颁发“山寨证书” 这家被点名的“五假”公司仍在营业 近期,江苏英才职业技能鉴定有限责任公司(以下简称英才公司)以“JYPC全国职业资格考试认证中心”名义颁发各类“山寨证书”被人力资源... -
 每日看点!哈弗酷狗内饰官图发布:坦克300同款档杆瞩目 哈弗酷狗内饰官图发布:坦克300同款档杆瞩目
每日看点!哈弗酷狗内饰官图发布:坦克300同款档杆瞩目 哈弗酷狗内饰官图发布:坦克300同款档杆瞩目 -
 新消息丨6月22日 江苏新增境外输入无症状感染者1例 据江苏省卫健委通报,6月22日0-24时,江苏无新增本土确诊病例,无新增本土无症状感染者。新增境外输入无症状感染者1例。目前,在定点医院隔离
新消息丨6月22日 江苏新增境外输入无症状感染者1例 据江苏省卫健委通报,6月22日0-24时,江苏无新增本土确诊病例,无新增本土无症状感染者。新增境外输入无症状感染者1例。目前,在定点医院隔离 -
 今日热讯:新一代本田雅阁谍照首曝:或将用上思域同款内饰 新一代本田雅阁谍照首曝:或将用上思域同款内饰
今日热讯:新一代本田雅阁谍照首曝:或将用上思域同款内饰 新一代本田雅阁谍照首曝:或将用上思域同款内饰 -
 每日热闻!官方平台出售事故车被罚10万!特斯拉:我们也不知道有事故 官方平台出售事故车被罚10万!特斯拉:我们也不知道有事故
每日热闻!官方平台出售事故车被罚10万!特斯拉:我们也不知道有事故 官方平台出售事故车被罚10万!特斯拉:我们也不知道有事故 -
 今日快讯:微软Win11 Dev预览版25145发布
IT之家6月23日消息,微软今天面向Dev开发频道发布了Windows11预览版25145系统更新。此版本推出了一些新功能,包括讲述人盲文
今日快讯:微软Win11 Dev预览版25145发布
IT之家6月23日消息,微软今天面向Dev开发频道发布了Windows11预览版25145系统更新。此版本推出了一些新功能,包括讲述人盲文 -
 荟萃群“英” “语”时俱进|深圳市弘金地学校学科交流会成功举行 6月7日,由深圳市坪山区弘金地学校举办、坪山区外国语文源学校协办的小学英语学科关于新课标背景下主教材与绘本融合的模式探究分享交流会在
荟萃群“英” “语”时俱进|深圳市弘金地学校学科交流会成功举行 6月7日,由深圳市坪山区弘金地学校举办、坪山区外国语文源学校协办的小学英语学科关于新课标背景下主教材与绘本融合的模式探究分享交流会在 -
 焦点日报:贵州打造现代农业产业技术体系 “黔芋7号”填补了贵州国审马铃薯品种的空白;“黔油早1号”“黔油早2号”解决了贵州油菜生产茬口矛盾;“油研50”成为长江流域市场油菜...
焦点日报:贵州打造现代农业产业技术体系 “黔芋7号”填补了贵州国审马铃薯品种的空白;“黔油早1号”“黔油早2号”解决了贵州油菜生产茬口矛盾;“油研50”成为长江流域市场油菜... - 【聚看点】西宁市应急局开展成品油市场“双随机一公开”执法检查 为进一步加强成品油市场管理,规范成品油经营秩序,及时消除安全隐患,遏制重特大事故发生,根据省政府办公厅《2022年成品油市场“双随...
-
 加密市场进入寒冬,下跌原因找到了 高通胀的市场环境下,激进加息如期而至。投资者恐慌指数暴涨,风险偏好与流动性的改变,导致风险资产迎来大规模的抛售,加密资产作为风险资
加密市场进入寒冬,下跌原因找到了 高通胀的市场环境下,激进加息如期而至。投资者恐慌指数暴涨,风险偏好与流动性的改变,导致风险资产迎来大规模的抛售,加密资产作为风险资 - 微资讯!宝马M3旅行版 宝马M3旅行版
-
 简讯:iOS 16 Beta 2 新增支持通过 LTE 网络备份
IT之家6月23日消息,今天苹果推送了iOS16 iPadOS16开发者预览版Beta2更新。根据苹果的更新发布说明,iOS16B
简讯:iOS 16 Beta 2 新增支持通过 LTE 网络备份
IT之家6月23日消息,今天苹果推送了iOS16 iPadOS16开发者预览版Beta2更新。根据苹果的更新发布说明,iOS16B -
 每日热闻!陕西西安学区划分方案出炉 公办学校实施单校划片 6月22日,西安市各区县、开发区公布了《2022年义务教育学校招生入学工作实施方案》和学区划分方案。方案明确,西安市公办学校的学区划分由各区
每日热闻!陕西西安学区划分方案出炉 公办学校实施单校划片 6月22日,西安市各区县、开发区公布了《2022年义务教育学校招生入学工作实施方案》和学区划分方案。方案明确,西安市公办学校的学区划分由各区 - 播报:6月22日辽宁丹东新增5例本土无症状感染者 央广网沈阳6月23日消息(见习记者麦丰)来自辽宁省卫生健康委员会的消息:6月22日0-24时,辽宁省无新增新冠肺炎确诊病例;新增5例本土无症状感
-
 每日焦点!上海22日本土确诊病例治愈出院9例、解除医学观察32例 记者从上海市卫健委获悉,6月22日0—24时,上海本土确诊病例治愈出院9例,解除医学观察本土无症状感染者32例。此外,2月26日0时至2022年6月22日24时
每日焦点!上海22日本土确诊病例治愈出院9例、解除医学观察32例 记者从上海市卫健委获悉,6月22日0—24时,上海本土确诊病例治愈出院9例,解除医学观察本土无症状感染者32例。此外,2月26日0时至2022年6月22日24时 -
 当前关注:高温黄色预警!河南等6省区最高气温达40℃以上 中央气象台6月23日06时继续发布高温黄色预警:预计,6月23日白天,新疆北部和南疆盆地、内蒙古西部、甘肃西部、华北中南部、黄淮北部、陕西中
当前关注:高温黄色预警!河南等6省区最高气温达40℃以上 中央气象台6月23日06时继续发布高温黄色预警:预计,6月23日白天,新疆北部和南疆盆地、内蒙古西部、甘肃西部、华北中南部、黄淮北部、陕西中 - 速递!河南省预计23日东部和南部局部有暴雨大暴雨 记者从河南省气象台了解到,6月22日14时-23日6时,河南省中西部、北部出现雨量分布不均的阵雨、雷阵雨,其中濮阳、安阳、鹤壁、三门峡、洛阳、
-
 每日快看:南京地铁回应女子手机开外放吃罚单:只是告知单 南京地铁回应女子手机开外放吃罚单:只是告知单
每日快看:南京地铁回应女子手机开外放吃罚单:只是告知单 南京地铁回应女子手机开外放吃罚单:只是告知单 -
 每日关注!5G云代驾 安全员远程接管无人驾驶汽车 近日,北青报记者来到百度公司,驾驶员正远程操控一辆奔跑在亦庄的无人驾驶汽车。驾驶舱的屏幕上,实时回传着车辆周边的环境画面和传感...
每日关注!5G云代驾 安全员远程接管无人驾驶汽车 近日,北青报记者来到百度公司,驾驶员正远程操控一辆奔跑在亦庄的无人驾驶汽车。驾驶舱的屏幕上,实时回传着车辆周边的环境画面和传感... -
 每日热门:山西商品房预售资金监管协议将按照示范文本签订 预售房资金被挪用,不能按时交房,甚至购房者积攒半辈子的积蓄被无良开发商拿钱跑路,针对近年来房地产市场出现的新情况和新挑战,近日...
每日热门:山西商品房预售资金监管协议将按照示范文本签订 预售房资金被挪用,不能按时交房,甚至购房者积攒半辈子的积蓄被无良开发商拿钱跑路,针对近年来房地产市场出现的新情况和新挑战,近日... - 【报资讯】日企感叹中国无人驾驶“领先好几步” 参考消息网6月21日报道据日本时事社6月19日报道,在全球范围内自动驾驶技术研发竞争日趋激烈的背景下,“无人出租车”在中国的商业化进...
-
 聚焦:甘肃高考成绩6月23日14时公布 (记者邸文炯)记者从甘肃省教育考试院获悉,2022年甘肃省普通高校招生全国统一考试成绩将于今日14时公布,考生可通过以下途径查询。1 电话查
聚焦:甘肃高考成绩6月23日14时公布 (记者邸文炯)记者从甘肃省教育考试院获悉,2022年甘肃省普通高校招生全国统一考试成绩将于今日14时公布,考生可通过以下途径查询。1 电话查 -
 精选!北京6月22日新增3例本土确诊病例 均住经开区 据北京市卫健委通报,6月22日0时至24时,北京新增3例本土确诊病例,无新增疑似病例和无症状感染者;新增1例境外输入确诊病例和1例无症状感染者
精选!北京6月22日新增3例本土确诊病例 均住经开区 据北京市卫健委通报,6月22日0时至24时,北京新增3例本土确诊病例,无新增疑似病例和无症状感染者;新增1例境外输入确诊病例和1例无症状感染者 -
 每日热议!聚资源凝合力 海河实验室创新联合体正式成立 6月22日,海河实验室发展论坛暨创新联合体成立大会在天津万丽宾馆隆重召开,百余家创新联合体成员单位的代表齐聚一堂,共同见证这一盛事...
每日热议!聚资源凝合力 海河实验室创新联合体正式成立 6月22日,海河实验室发展论坛暨创新联合体成立大会在天津万丽宾馆隆重召开,百余家创新联合体成员单位的代表齐聚一堂,共同见证这一盛事... -
 微头条丨迪卡侬发布概念电动自行车:支持无线解锁 续航90千米 迪卡侬发布概念电动自行车:支持无线解锁续航90千米
微头条丨迪卡侬发布概念电动自行车:支持无线解锁 续航90千米 迪卡侬发布概念电动自行车:支持无线解锁续航90千米 -
 观速讯丨【央广网评】让网络主播在“规则”轨道内跑得更远、更好 6月22日,国家广播电视总局、文化和旅游部联合发布《网络主播行为规范》(以下简称《规范》),明确提出网络主播在提供网络表演及视听节...
观速讯丨【央广网评】让网络主播在“规则”轨道内跑得更远、更好 6月22日,国家广播电视总局、文化和旅游部联合发布《网络主播行为规范》(以下简称《规范》),明确提出网络主播在提供网络表演及视听节... -
 【热闻】天津市宝坻区燃气泄漏爆燃事故7名伤员出院 9名涉事责任人被控制 6月22日,天津市“6·21”燃气泄漏爆燃事故调查组通报事故最新情况,内容如下:天津市宝坻区燃气泄漏爆燃事故发生后,市、区两级卫健部...
【热闻】天津市宝坻区燃气泄漏爆燃事故7名伤员出院 9名涉事责任人被控制 6月22日,天津市“6·21”燃气泄漏爆燃事故调查组通报事故最新情况,内容如下:天津市宝坻区燃气泄漏爆燃事故发生后,市、区两级卫健部... -
 每日热闻!天津海关十年来共侦破毒品走私案件165起 “6 26”国际禁毒日前夕,天津海关对外发布,自党的十八大以来,天津海关始终保持打击毒品走私高压态势,共侦破毒品走私案件165起,查...
每日热闻!天津海关十年来共侦破毒品走私案件165起 “6 26”国际禁毒日前夕,天津海关对外发布,自党的十八大以来,天津海关始终保持打击毒品走私高压态势,共侦破毒品走私案件165起,查... -
 快资讯丨余承东大赞舒适度超越所有豪车!问界M7实车曝光 余承东大赞舒适度超越所有豪车!问界M7实车曝光
快资讯丨余承东大赞舒适度超越所有豪车!问界M7实车曝光 余承东大赞舒适度超越所有豪车!问界M7实车曝光 -
 【报资讯】苹果 watchOS 9 开发者预览版 Beta 2 发布
【点此直达描述文件下载】IT之家6月23日消息,苹果今日向AppleWatch用户推送了watchOS9开发者预览版Beta2更新(
【报资讯】苹果 watchOS 9 开发者预览版 Beta 2 发布
【点此直达描述文件下载】IT之家6月23日消息,苹果今日向AppleWatch用户推送了watchOS9开发者预览版Beta2更新( -
 时讯:苹果 macOS 13 开发者预览版 Beta 2 发布
IT之家6月23日消息,苹果今日向Mac电脑用户推送了macOS13开发者预览版Beta2更新(内部版本号:22A5286j),本次更新
时讯:苹果 macOS 13 开发者预览版 Beta 2 发布
IT之家6月23日消息,苹果今日向Mac电脑用户推送了macOS13开发者预览版Beta2更新(内部版本号:22A5286j),本次更新 -
 每日热议!比混动丰田还省 吉利帝豪醇电版上市:12.98万 比混动丰田还省吉利帝豪醇电版上市:12 98万
每日热议!比混动丰田还省 吉利帝豪醇电版上市:12.98万 比混动丰田还省吉利帝豪醇电版上市:12 98万 - 即时看!车企APP积分被当“羊毛”转卖:月入十万成本几乎为零 车企APP积分被当“羊毛”转卖:月入十万成本几乎为零
- 动态:大型龙江剧《萧红》在哈尔滨首演 6月22日,由黑龙江省文化和旅游厅主办、省龙江剧艺术中心演出的国家艺术基金资助项目、大型原创龙江剧《萧红》在哈尔滨首演。大型原创龙...
热门资讯
-
 知名数字资产交易平台8V上线模拟交易,零成本体验合约交易 近日,全球知名元宇宙生态数字资产...
知名数字资产交易平台8V上线模拟交易,零成本体验合约交易 近日,全球知名元宇宙生态数字资产... -
 刘桐树主任医师 ——复兴中医养生健康的先驱者 看的是病,救的是心,开的是药,给的...
刘桐树主任医师 ——复兴中医养生健康的先驱者 看的是病,救的是心,开的是药,给的... -
 用颜层®钝针填充太阳穴,给你意想不到的效果 从美的角度来讲,相比曲折的脸部线...
用颜层®钝针填充太阳穴,给你意想不到的效果 从美的角度来讲,相比曲折的脸部线... -
 李亚四繁殖场:从崛起到衰败,曾是龙鱼界大佬,如今却面临欠租倒闭 李亚四,一位经常穿着白色衬衫,谦虚...
李亚四繁殖场:从崛起到衰败,曾是龙鱼界大佬,如今却面临欠租倒闭 李亚四,一位经常穿着白色衬衫,谦虚...
观察
图片新闻
-
 网红服装店“超长预售”惹争议 时间成本完全转嫁给消费者 发货太慢了!是下单了才去种棉花吗?...
网红服装店“超长预售”惹争议 时间成本完全转嫁给消费者 发货太慢了!是下单了才去种棉花吗?... -
 金融服务实体经济质效不断提升 稳定宏观经济大盘 昨天,中国人民银行发布今年第一季...
金融服务实体经济质效不断提升 稳定宏观经济大盘 昨天,中国人民银行发布今年第一季... -
 服务贸易取得“开门稳” 保障外贸产业链供应链稳定畅通 当前我国外贸发展的环境更趋严峻复...
服务贸易取得“开门稳” 保障外贸产业链供应链稳定畅通 当前我国外贸发展的环境更趋严峻复... -
 多个城市因城施策 促房地产市场平稳健康发展 今年以来,为落实中央支持合理住房...
多个城市因城施策 促房地产市场平稳健康发展 今年以来,为落实中央支持合理住房...
精彩新闻
-
 每日焦点!油价创史上新高!美国出“奇招”:为省油减少警车巡逻 油价创史上新高!美国出“奇招”:...
每日焦点!油价创史上新高!美国出“奇招”:为省油减少警车巡逻 油价创史上新高!美国出“奇招”:... - 每日观点:七台河被授予“奥运冠军之城”纪念奖杯 为表彰在北京2022年冬奥会上为中国...
-
 滚动:重庆市气象局发布暴雨黄色预警(Ⅲ级/较重) 重庆市气象台2022年6月22日21时40...
滚动:重庆市气象局发布暴雨黄色预警(Ⅲ级/较重) 重庆市气象台2022年6月22日21时40... -
 【新视野】苹果 iOS/iPadOS 16 开发者预览版 Beta 2 发布
【点此直达描述文件下载】IT之家6...
【新视野】苹果 iOS/iPadOS 16 开发者预览版 Beta 2 发布
【点此直达描述文件下载】IT之家6... - 前沿热点:《王者荣耀》S28赛季 “沙海飞舟”更新,新英雄戈娅上线
IT之家6月22日消息,《王者荣耀》...
- 微速讯:法拉利告别内燃机 法拉利告别内燃机
-
 新资讯:AMD 上线“GPU 比较工具”,可在不同游戏中与英伟达型号对比
IT之家6月22日消息,据外媒报道,A...
新资讯:AMD 上线“GPU 比较工具”,可在不同游戏中与英伟达型号对比
IT之家6月22日消息,据外媒报道,A... - 今日看点:山东理工大学与博仲医药签订合作协议 6月21日,山东理工大学、博仲医药...
- 当前热点-哈尔滨:全方位推动生物经济高质量发展 6月22日,哈尔滨市政府新闻办组织...
- 邵阳、武冈房屋安全鉴定找第三方检测机构的正规军中国有色金属长勘院 安得广厦千万间,大庇天下寒士俱欢...
-
 传祺M8“棕山云海”四座版,带你舒适进击珠峰 许多人没有去过西藏,但在他们的心...
传祺M8“棕山云海”四座版,带你舒适进击珠峰 许多人没有去过西藏,但在他们的心... -
 庆祝中日邦交正常化50周年,陕西民俗文化节将在东京举办 2022陕西民俗文化节将于6月20日在...
庆祝中日邦交正常化50周年,陕西民俗文化节将在东京举办 2022陕西民俗文化节将于6月20日在... - 微速讯:提单是什么?快递提单是什么? 提单是什么(快递提单是什么)***请...
- 解锁消费新体验!健合旗下品牌618多平台斩获第一 线上消费盛宴618,一直被业界视为...
-
 今日报丨后妃等级怎么划分的?古代妃子位分顺序表 后妃等级(古代妃子位分顺序表) ...
今日报丨后妃等级怎么划分的?古代妃子位分顺序表 后妃等级(古代妃子位分顺序表) ... -
 每日热议!今日山西钢材最新价?钢材西本每日报价多少?
每日热议!今日山西钢材最新价?钢材西本每日报价多少? -
 前沿资讯!德阳安医生事件处理结果是什么? 近日德阳安医生事件处理结果登录了...
前沿资讯!德阳安医生事件处理结果是什么? 近日德阳安医生事件处理结果登录了... - 观焦点:mac版是什么意思?mac是平板的意思吗? mac版是什么意思(mac是平板的意思...
- 观热点:mhdd硬盘检测工具有哪些?pc3000硬盘修复教程 mhdd硬盘检测工具(pc3000硬盘修复...
-
 微信微粒贷怎么开通?微信备用金2000元申请额度入口 微信微粒贷怎么开通?微信备用金200...
微信微粒贷怎么开通?微信备用金2000元申请额度入口 微信微粒贷怎么开通?微信备用金200... -
 什么叫央票?与国债的区别?央票和国债的区别? 什么叫央票?与国债的区别?央票和国...
什么叫央票?与国债的区别?央票和国债的区别? 什么叫央票?与国债的区别?央票和国... - 早餐一杯维他奶豆奶 更多人的优质蛋白首选 黄豆是豆科植物大豆的黄色种子,被...
- 百事通!孟州在哪里?河南孟州穷吗? 孟州在哪里(河南孟州穷吗)焦作市是...
-
 当前信息:格力空调怎么样?格力空调润慧是高端机吗? 格力空调怎么样(格力空调润慧是高...
当前信息:格力空调怎么样?格力空调润慧是高端机吗? 格力空调怎么样(格力空调润慧是高... - 看点:湖南师范大学怎么样?湖南师范大学有多厉害? 湖南师范大学怎么样(湖南师范大学...
-
 股票杠杆可以加多少倍?10万10倍杠杆能挣多少? 股票杠杆可以加多少倍?10万10倍杠...
股票杠杆可以加多少倍?10万10倍杠杆能挣多少? 股票杠杆可以加多少倍?10万10倍杠... -
 股票杠杆最多可以多少倍?杠杆股票交易类型详细介绍 股票杠杆最多可以多少倍?杠杆股票...
股票杠杆最多可以多少倍?杠杆股票交易类型详细介绍 股票杠杆最多可以多少倍?杠杆股票... -
 杠杆炒股是什么意思?炒股最大杠杆是多少倍? 杠杆炒股是什么意思?炒股最大杠杆...
杠杆炒股是什么意思?炒股最大杠杆是多少倍? 杠杆炒股是什么意思?炒股最大杠杆... -
 中国重汽VGV VX7 与您解锁精致露营 现代生活方式主要是由网络和通信,...
中国重汽VGV VX7 与您解锁精致露营 现代生活方式主要是由网络和通信,... -
 pb是什么意思?股票pb指标高好还是低好? pb是什么意思?pb在股票领域是指平...
pb是什么意思?股票pb指标高好还是低好? pb是什么意思?pb在股票领域是指平... -
 德华安顾人寿启动空中客服,提升保险温度 在疫情的倒逼下,各行各业一系列云...
德华安顾人寿启动空中客服,提升保险温度 在疫情的倒逼下,各行各业一系列云... -
 汐羽老师—以美丽改变人生为己任 汐羽老师说:现在化妆造型师很多,很...
汐羽老师—以美丽改变人生为己任 汐羽老师说:现在化妆造型师很多,很... -
 基金份额越多是不是收益就越多?持有金额和持有份额的区别是什么? 基金市场瞬息万变,那么基金份额越...
基金份额越多是不是收益就越多?持有金额和持有份额的区别是什么? 基金市场瞬息万变,那么基金份额越... -
 回乡还是坚守?Soul App上年轻人有话说 随着毕业季的到来,Soul广场上悄然...
回乡还是坚守?Soul App上年轻人有话说 随着毕业季的到来,Soul广场上悄然... - 海汽集团拟收购海旅免税100%股权 主业转型综合旅游 6月21日,海汽集团公告,经初步预...
- 2022“国际瑜伽日”系列官方活动圆满落幕,由印度大使馆、印梵西瓦瑜伽共同主办,... 近日,由印度大使馆、印梵西瓦瑜伽...
-
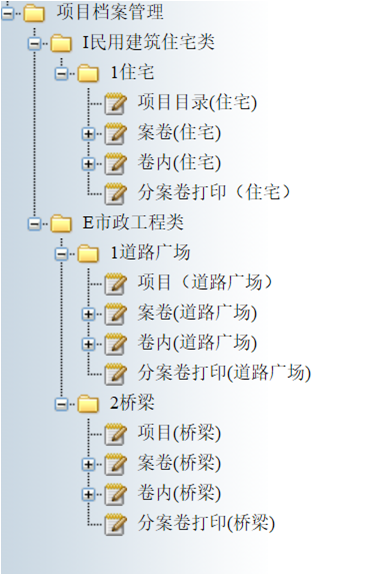 勤哲Excel服务器无代码实现工程档案管理系统 如今,随着我国信息水平的不断提高...
勤哲Excel服务器无代码实现工程档案管理系统 如今,随着我国信息水平的不断提高... -
 河南“小麦大蒜”抵首付 地产商的营销动作花样百出 楼市低温、房子难卖,地产商的营销...
河南“小麦大蒜”抵首付 地产商的营销动作花样百出 楼市低温、房子难卖,地产商的营销... - 阿里健康京东健康股价跌超9% 第三方平台将禁止自营药品? 6月22日上午,阿里健康(00241 HK)...
-
 中央空调、有氧健身、AI保镖,现代农产品的生活你想象不到 你见过五星级待遇的鸡吗?你吃过健...
中央空调、有氧健身、AI保镖,现代农产品的生活你想象不到 你见过五星级待遇的鸡吗?你吃过健... -
 上海中小微企业纾困政策有最新进展 政策细则加快落地 作为稳经济、稳市场、稳就业的主体...
上海中小微企业纾困政策有最新进展 政策细则加快落地 作为稳经济、稳市场、稳就业的主体... - 理想汽车正式发布理想L9 理想汽车第二款量产车 6月21日,理想汽车正式发布理想L9...
-
 坊间掀起一波“微醺热” 新酒饮产品愈发丰富 坊间掀起了一波微醺热。6月21日,...
坊间掀起一波“微醺热” 新酒饮产品愈发丰富 坊间掀起了一波微醺热。6月21日,... -
 新东方在线的股价大幅波动 腾讯大额减持套现 东方甄选和新东方在线的动态仍在引...
新东方在线的股价大幅波动 腾讯大额减持套现 东方甄选和新东方在线的动态仍在引... - 乖宝宠物更新招股说明书 IPO审核状态进入问询阶段 冲刺IPO,乖宝宠物掘金宠物赛道。6...
- 认养一头牛完成上市辅导 具备进入证券市场的基本条件 日前,浙江证监局公示了中信证券《...
-
 IFSM国际时尚超模大赛 深圳观澜湖华丽绽放 2022年6月18日,由北京三阳丽景国...
IFSM国际时尚超模大赛 深圳观澜湖华丽绽放 2022年6月18日,由北京三阳丽景国... - 多家品牌商家618理性上新 下沉市场获青睐 6·18喧闹过后,品牌商们忙着复盘...
- 银保监会对村镇银行开出121张罚单 119家村镇银行挨罚 近期,河南多家村镇银行提现难事件...
- “东方甄选”直播间热度持续走高 新东方在线股价涨幅超300% 东方甄选和新东方在线的动态仍在引...